DNA Methylation Profiling for Translational Discovery
Epigenome Technologies provides end-to-end DNA methylation services, from gold-standard WGBS to bisulfite-free TAPS+/5-base sequencing. We align method selection to your experimental design deliver single-base resolution methylation maps ready for downstream analysis. We also combine chromatin profiling with DNA methylation to provide high-content epigenetic readouts.
Gold Standard
WGBS
5mC -> T
TAPS
Chromatin + 5mC
CUT&TAPS
Choose the right methylation profiling method
| Service | Cat. No | Input | Modification Detected | Best For | Inquiry |
|---|---|---|---|---|---|
| WGBS | EGT-WG-401 | 50+ ng DNA | 5mC (CpG, CHG, CHH) | Genome-wide 5mC maps, reference datasets, aging and cancer cohorts | Request Quote |
| TAPS | EGT-TP-502 | 1ng DNA | 5mC (CpG, CHG, CHH) | cfDNA, FFPE and scarce samples, full fragments for long reads | Request Quote |
| CUT&TAPS | EGT-CTAP-603 | 50,000 nuclei or fewer | 5mC at antibody-defined loci | Low-cost, high-coverage joint histone/TF profiling with DNA methylation | Request Quote |
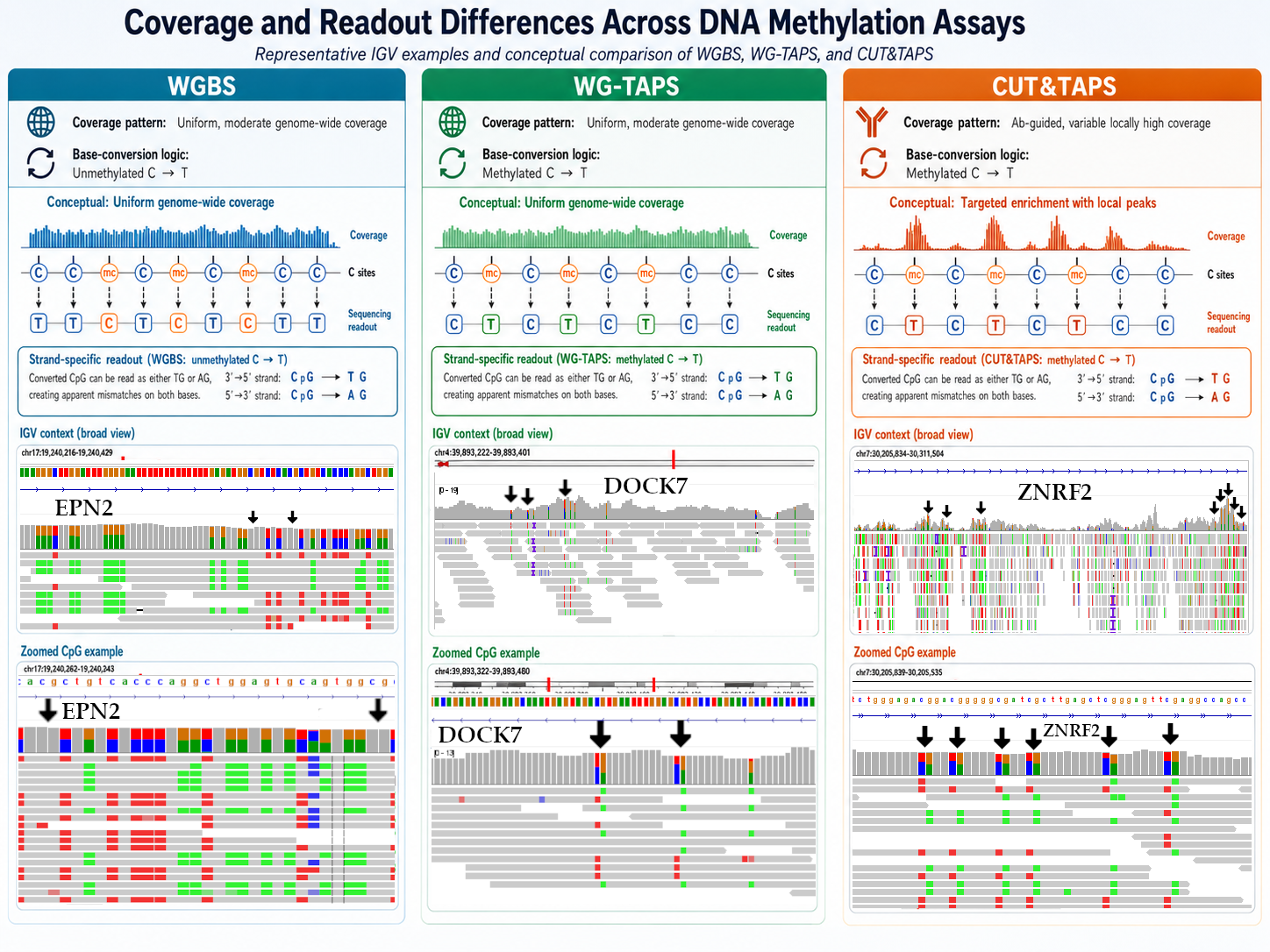
Single-base methylation profiling across sample types and scales
WGBS, TAPS, and CUT&TAPS map DNA methylation at single-base resolution across distinct biological scales. Epigenome Technologies can help match the technology to experimental questions and sample quantities, delivering best-in-class discovery-oriented methylation datasets.
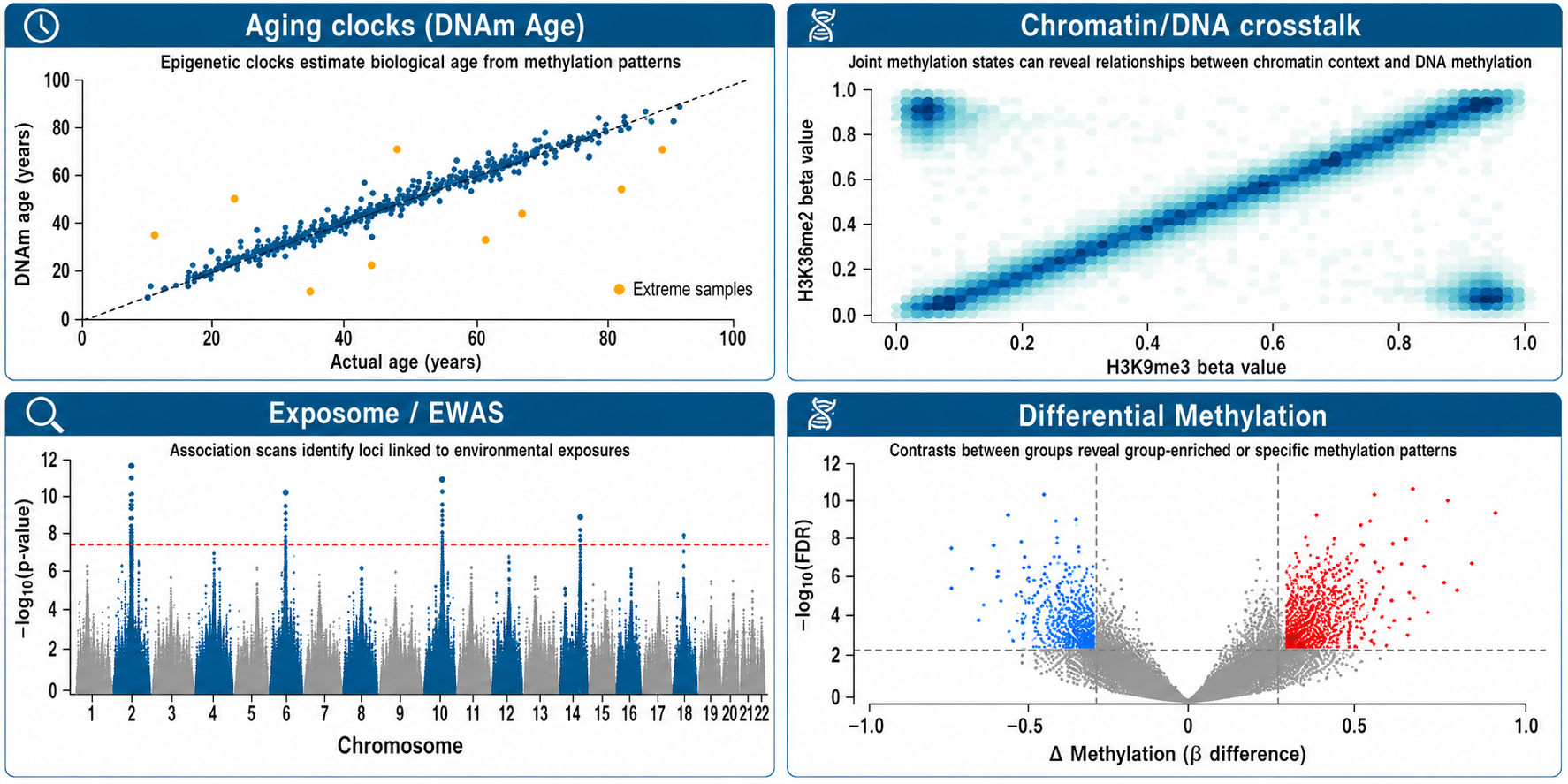
Common applications
- Cancer methylation landscape profiling and biomarker discovery
- Aging clock validation and epigenetic age acceleration studies
- Cell-free DNA / circulating tumor DNA methylation studies
- FFPE and archival specimen rescue for retrospective cohorts
- Chromatin-state-linked methylation at enhancers and promoters
- Chromatin/Methylation crosstalk and high-content epigenetics
Included with every engagement
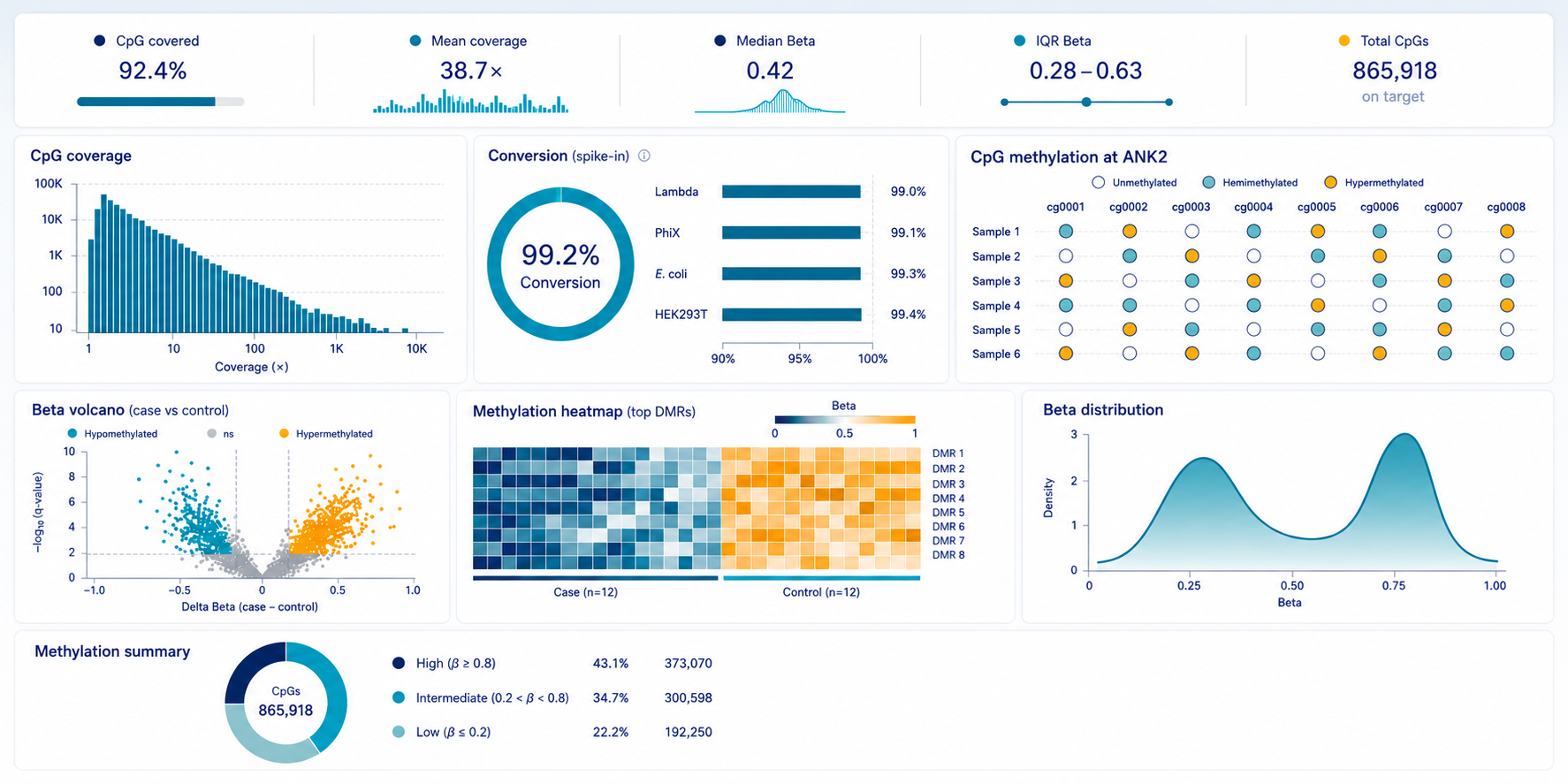
- Experimental design and assay alignment with sample availability and project goals
- DNA controls for %conversion normalization
- Quality control reviews at specified checkpoints
- End-to-end sequencing and data processing
- Optional integration with RNA-seq, ATAC-seq, or chromatin profiling datasets
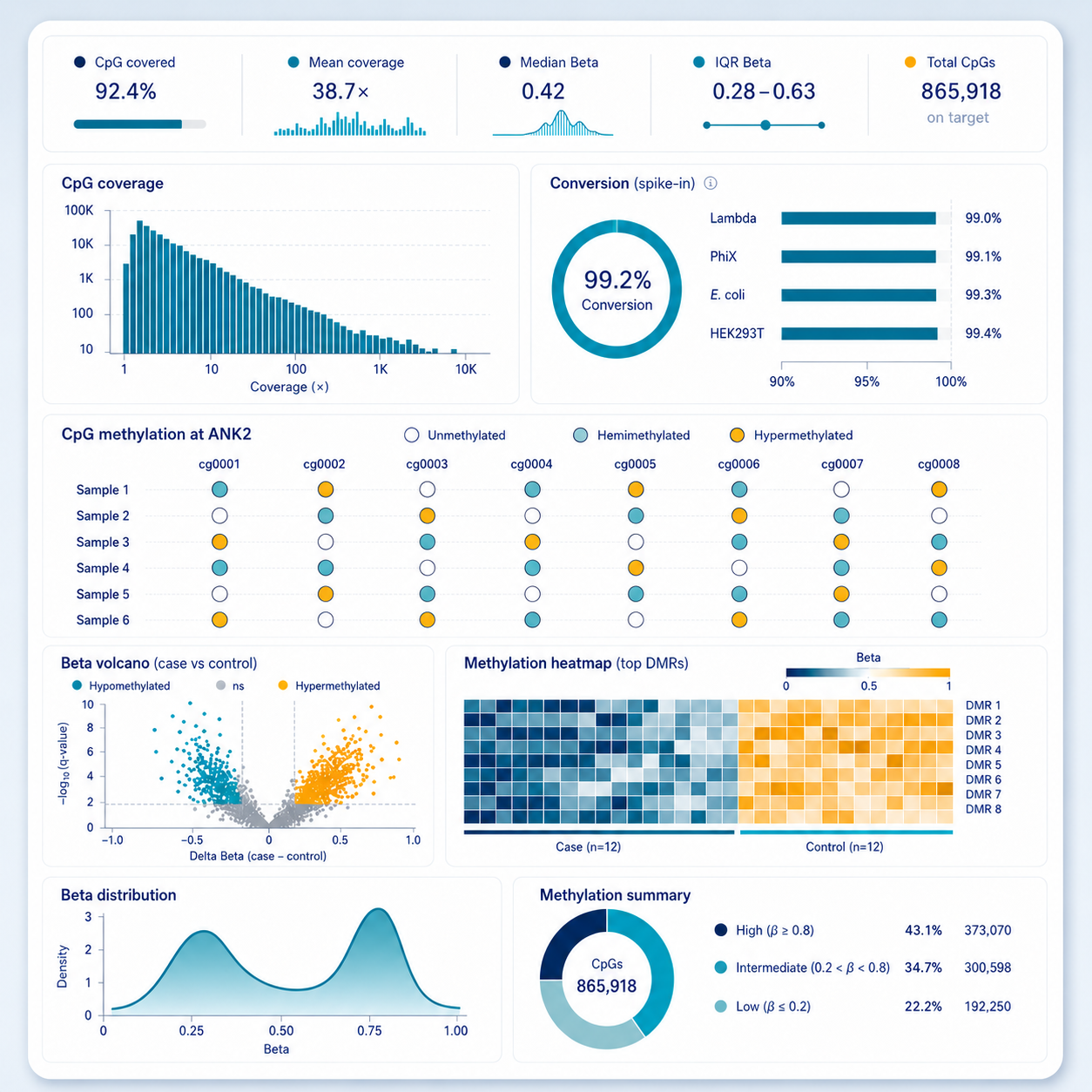
WGBS: Single-Base Genome-Wide Methylation Mapping
Whole genome bisulfite sequencing provides single-base resolution methylation calls across every CpG in the genome, establishing the reference standard for methylation landscape studies. Our WGBS service covers library prep, bisulfite conversion, sequencing, and Bismark-based methylation calling, delivering CpG-resolution bedGraph and CpG_report files ready for downstream DMR analysis.
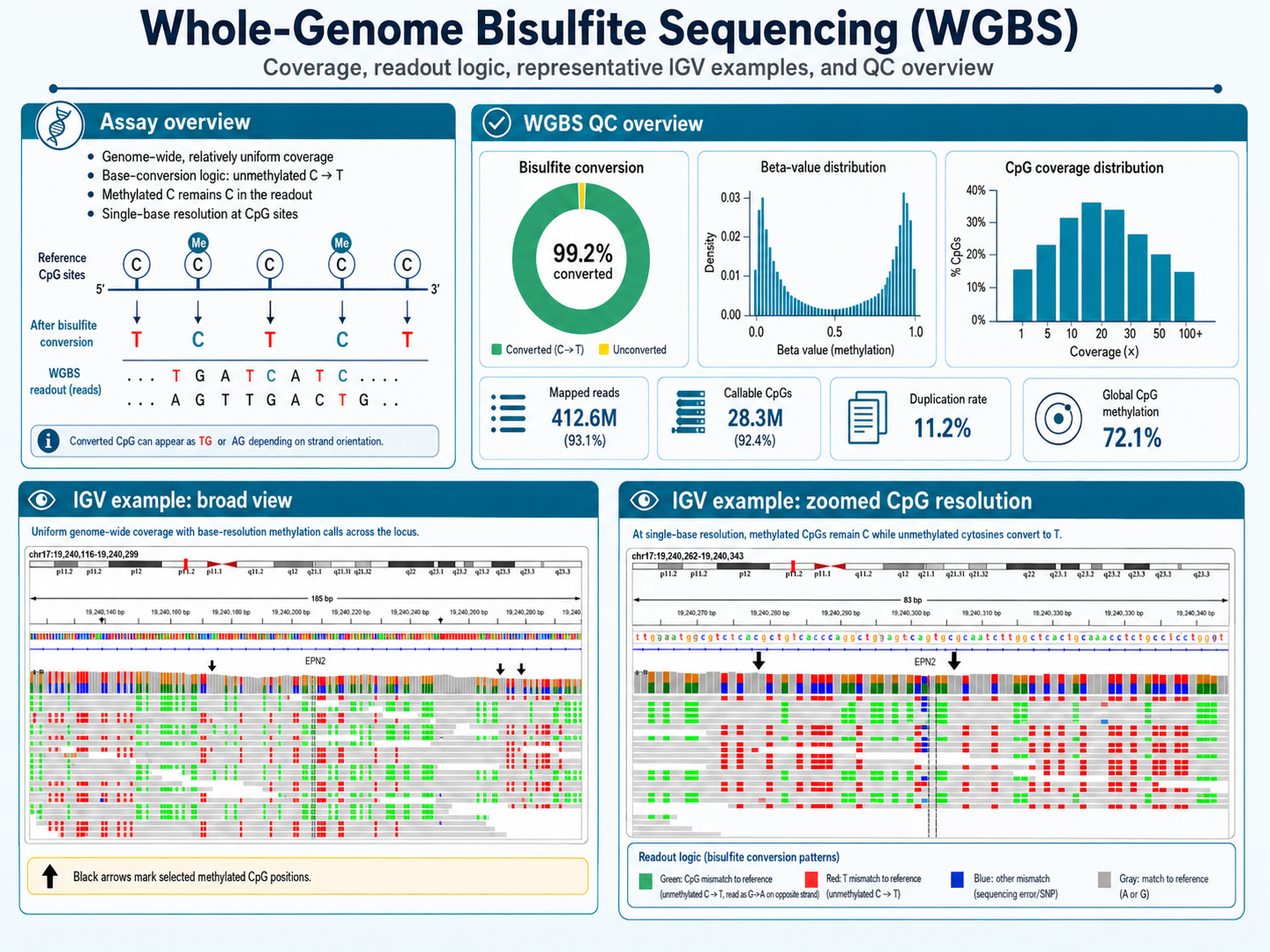
Best for
- High-input samples (≥100 ng genomic DNA)
- Reference-quality genome-wide 5mC maps
- Aging clock validation and cancer methylation landscapes
- Allele-specific methylation and imprinting studies
- Benchmark comparisons across cohorts and platforms
Genome-wide CpG Methylation
QC metrics
- Bisulfite conversion efficiency ≥ 99.5%
- Complexity sufficient for ≥ 100× median across target regions
- Replicate CpG correlation > 0.97
Workflow
-
Plan
Study design review covering coverage targets, cohort batching, spike-in strategy, and downstream analysis requirements.
-
Fragment & convert
DNA shearing to target fragment size, bisulfite treatment, and desulfonation with integrated conversion efficiency checkpoint.
-
Library construction
Adapter ligation, PCR amplification, and size selection with conversion QC gate prior to sequencing approval.
-
Sequence & call
Paired-end sequencing followed by Bismark alignment and CpG-resolution methylation calling across all three cytosine contexts.
Deliverables
- Library QC metrics including conversion efficiency and fragment distribution
- FASTQ files and Bismark-aligned BAM files
- CpG-resolution methylation calls (bedGraph and CpG_report formats)
- DMR analysis with annotated genomic context
- Detailed QC report with coverage distributions and duplication metrics
Advantages
- Single-base resolution across the entire CpG methylome
- Detects CpG, CHG, and CHH methylation contexts
- Gold-standard benchmark for inter-lab and multi-cohort comparisons
- Compatible with established aging clock and DMR pipelines
- Comprehensive coverage enables allele-specific methylation calling
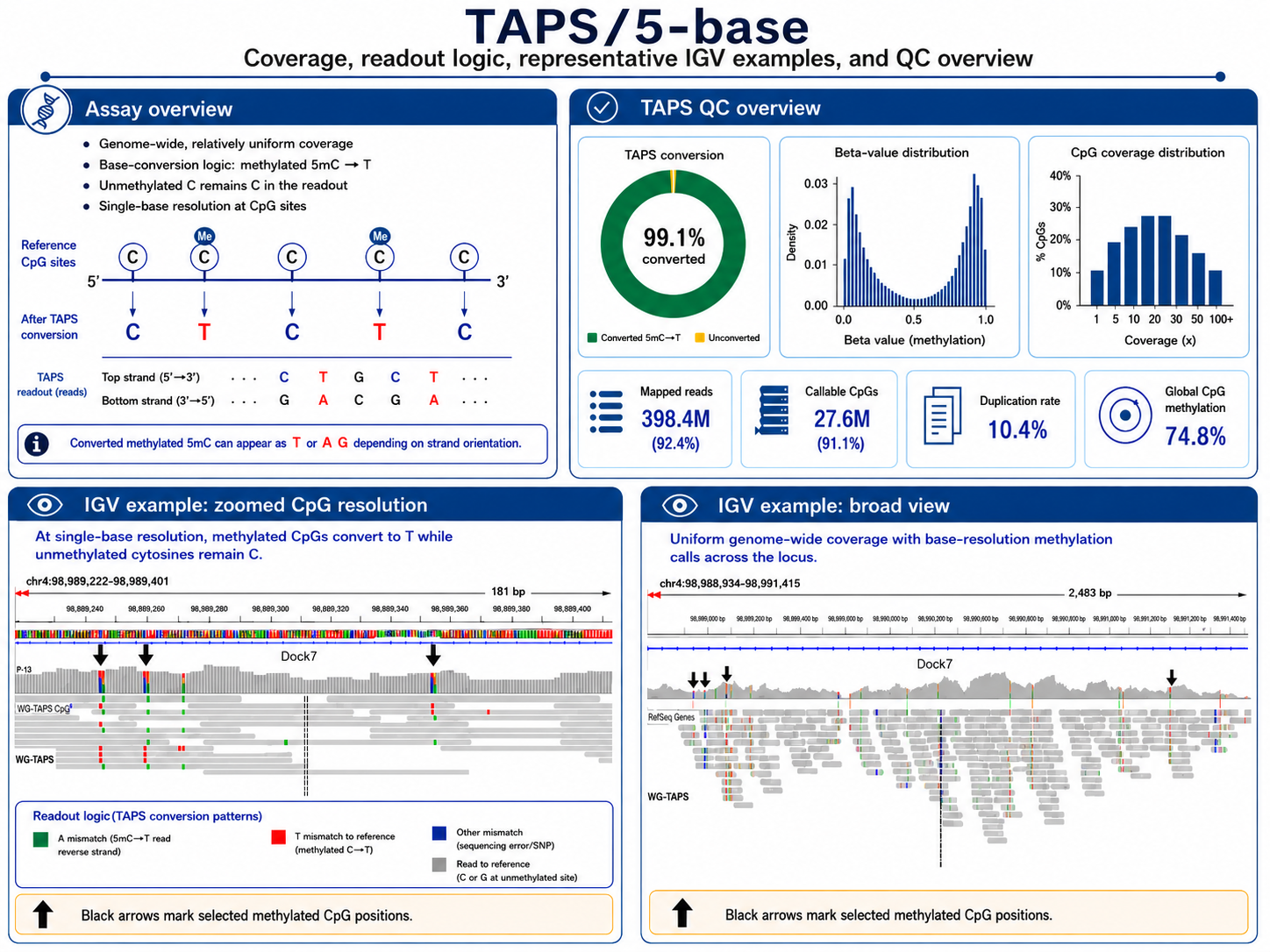
TAPS: Bisulfite-Free Resolution of 5mC
Confident application of Watchmaker TAPS+ or Illumina 5-base to convert 5mC into readable bases without bisulfite treatment, preserving DNA integrity and enabling lower-input workflows. TAPS+ and 5-base deamination kits are less damaging to DNA, and thus better able to retain large fragments for long-range methylation haplotyping.
Best for
- FFPE and archival specimens where bisulfite degradation limits data quality
- Low-input DNA (1 ng) with minimal fragmentation tolerance
- Fragmentomics data where fragment size is valuable
- Long-range methylation haplotyping
Bisulfite-Free Chemistry
QC metrics
- Conversion efficiency ≥ 98%
- CpG coverage ≥ 10× median at 1ng DNA
- Replicate CpG correlation (Pearson r) > 0.97
Workflow
-
Plan
Sample QC review, coverage targets, spike-in strategy, and confirmation of 5hmC enrichment regions of interest.
-
Methylation conversion
Bisulfite-free chemistry via TAPS+ or 5-base deamination preserves DNA integrity and reduces depurination artifacts.
-
Library construction
Adapter ligation, amplification, and size selection with chemistry efficiency QC gate prior to sequencing approval.
-
Sequence & call
Paired-end sequencing, TAPS-aware alignment, and generation of separate 5mC and 5hmC methylation call files at single-base resolution.
Deliverables
- Library QC metrics with efficiency reports
- FASTQ files and TAPS-aligned BAM files
- Methylation quantification at CpG and CHH
- QC report comparing 5mC and 5hmC profiles across samples
Advantages
- Bisulfite-free chemistry reduces DNA degradation and improves FFPE data quality
- Lower DNA input than WGBS (down to 1 ng)
- Cleaner base calls with reduced C→T noise from depurination
- Compatible with long-read sequencing for phased methylation analysis
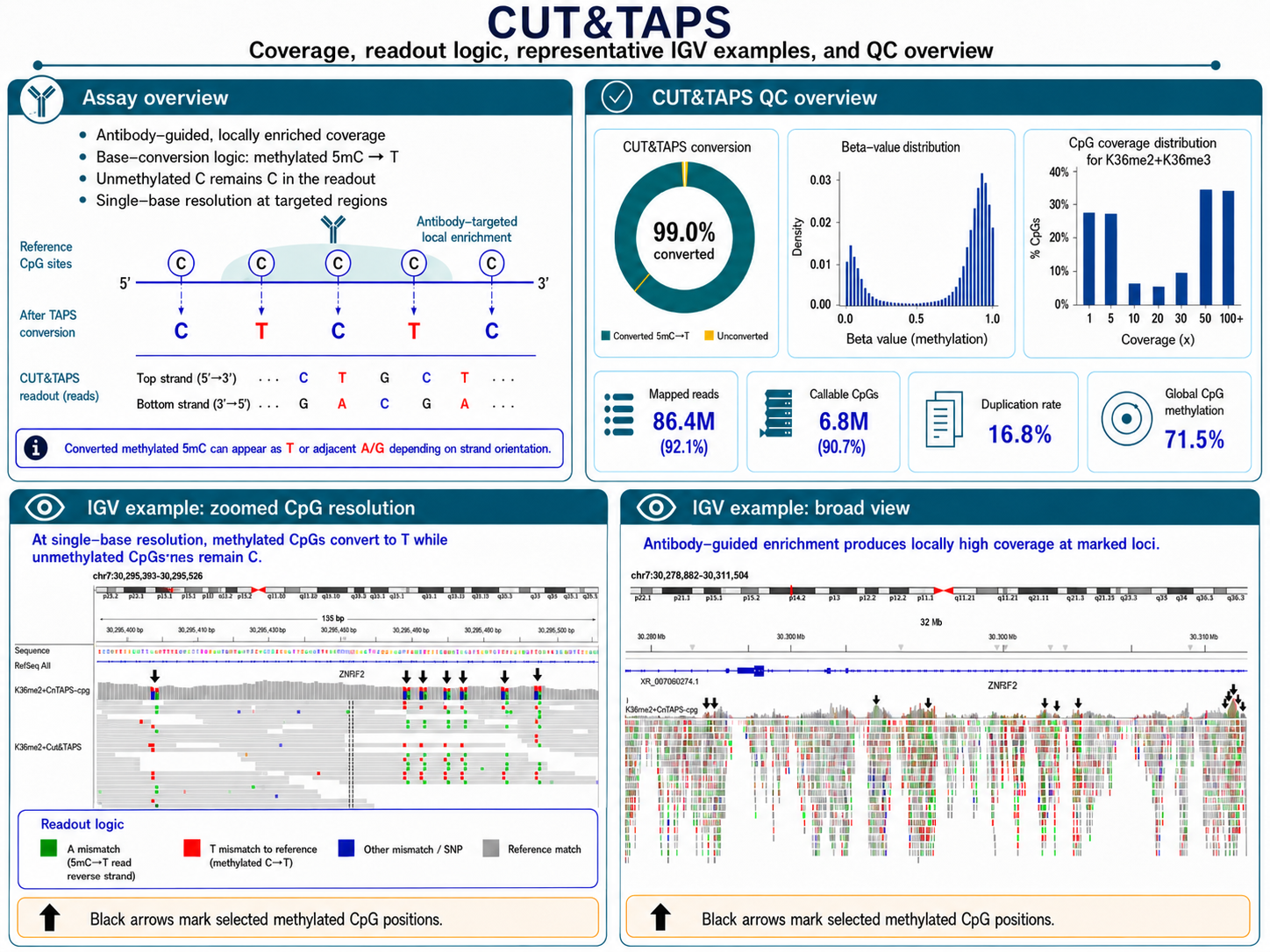
CUT&TAPS: Joint Chromatin/Methylation Profiling
CUT&TAPS combines CUT&Tag-style antibody-guided chromatin cleavage with bisulfite-free chemistry to map DNA methylation specifically at loci defined by a protein of interest — such as a histone modification, transcription factor, or methylation proteins themselves such as MECP2 or DNMT3. The approach delivers targeted methylation data from as few as 50,000 nuclei, without the sequencing cost of whole-genome coverage.
Best for
- Ultra-low-input samples (50,000 cells)
- Available in single-cell resolution
- Methylation profiling adjacent to specific histone marks or TF binding sites
- Methylation quantification at actively-methylated CpGs
- Chromatin-state-linked methylation in disease and development
- Studies requiring simultaneous epigenetic mark and methylation readout
- Single-cell methylation studies
Chromatin–Methylation Crosstalk
QC metrics
- Antibody enrichment confirmed by FRiP at known target loci ≥ 0.50
- TAPS chemistry efficiency verified by spike-in controls
- On-target CpG coverage ≥ 30× at enriched regions
- Replicate CpG correlation at enriched loci > 0.96
Workflow
-
Plan
Antibody validation, target locus strategy, cell input assessment, and spike-in design with your project scientist.
-
Antibody binding & cleavage
Cell permeabilisation, antibody incubation, protein A-MNase tethering, and on-bead cleavage at antibody-defined target loci.
-
Methylation conversion
TAPS+ conversion or 5-base deamination, depending on application, convert methylation to readnable bases
-
Library & analysis
Adapter ligation, sequencing, TAPS-aware alignment, and methylation calling within antibody-enriched windows.
Deliverables
- Library QC metrics with enrichment and TAPS chemistry efficiency reports
- FASTQ files and enrichment-aware aligned BAM files
- Methylation calls at CpG resolution within antibody-enriched regions
- Peak calls and coverage tracks showing antibody enrichment
- Integrated report linking chromatin mark occupancy to local methylation state
Advantages
- Combines CUT&Tag antibody precision with bisulfite-free methylation readout
- Ultra-low-input — suitable for primary cells and clinical specimens
- Single-cell compatible with Chromium or other platforms
- Focused sequencing depth: cost-efficient for targeted hypotheses
- Links histone modification state directly to DNA methylation at single-locus resolution
- No whole-genome coverage required — reduces compute and sequencing costs
Partner with our scientists
Share your sample type, modification targets, and cohort size. We will return a scoped methylation profiling brief outlining recommended method selection (WGBS, TAPS+, or CUT&TAPS), QC checkpoints, and downstream reporting.
- Sample requirements: Method-specific inputs from 50,000 nuclei (CUT&TAPS) to 1ng DNA (TAPS/5base), to low-cost high inputs (WGBS).
- Storage guidance: Fresh, cryopreserved, or FFPE submissions accepted with documented handling.
- Data options: Raw, processed, and analyzed outputs available individually or bundled.
- Support: Project scientists provide experimental planning and guidance, data reviews, and troubleshooting.