Perturb-seq Reveals an Compensation Mechanism Underpinning Resistance in AML
An exciting new study employs Perturb-seq to reveal an epigenetic compensation mechanism underpinning therapeutic resistance in acute myeloid leukemia.
Perturb-seq: Exploring the Activating and Repressive Epigenetic Forces Acting in Acute Myeloid Leukemia
While therapies targeting individual epigenetic regulators in lysine methyltransferase 2A (KMT2A; also known as MLL)-rearranged acute myeloid leukemia (KMT2A-r AML) - an aggressive subtype marked by a differentiation block and poor clinical outcomes - initially display anti-tumor efficacy, responses often remain incomplete and short-lived (Zehtabcheh et al.). Currently, research groups aim to determine if resistance arises from compensatory interactions between chromatin-modifying complexes and, if so, how these interactions converge on programs that control cell fate. Activating epigenetic complexes can drive oncogenic gene expression programs to promote self-renewal (Zehtabcheh et al., Yan et al., and Katsumoto et al.) while repressive epigenetic complexes can silence critical differentiation genes to lock cells in an immature state (Schurer et al.); however, the relationship between these activating and repressive forces remains highly context-dependent and poorly understood (Sheridan et al., Zhou et al., and Sparbier et al.). This incomplete understanding has slowed the design of rational combination therapies; could Perturb-seq - a single-cell CRISPR-based screening assay (Dixit et al.) - provide an opportunity to explore epigenetic cooperativity in KMT2A-r AML?
As described in a recent Leukemia study, researchers led by Rui Lu and Changde Cheng (University of Alabama at Birmingham) combined Perturb-seq with computational modeling to comprehensively map the transcriptional impact of perturbing critical epigenetic regulators to map the functional architecture of the KMT2A-r AML compensatory network at single-cell resolution and inform the development of precision combination therapies (Aryal et al.).
Paired-Tag technology from Epigenome Technologies generates joint epigenetic and transcriptomic profiles at single-cell resolution and detects histone modifications and RNA transcripts in nuclei with efficiency comparable to single-nucleus RNA-seq/ChIP-seq assays. Could an in-depth analysis of histone modifications and transcriptomic profiles of the same single cells via the integration of Paired-Tag reveal more regarding the epigenetic compensation mechanisms that underpin therapeutic resistance in KMT2A-r AML?
An Epigenetic Mechanism that Represses Differentiation in Acute Myeloid Leukemia Revealed by Perturb-seq
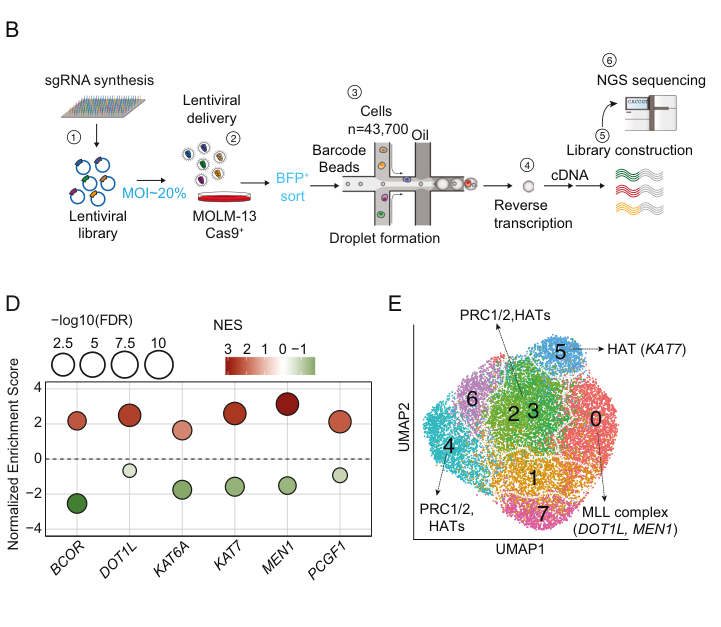
The basis of this study involved high-throughput Perturb-seq assays targeting sixteen epigenetic regulators selected for their established roles in chromatin modification and transcriptional regulation in a KMT2A-r AML cell line. This approach, when applied alongside integrated computational modeling, revealed a synergistic hub comprising the histone acetyltransferase KAT6A, the chromatin adaptor protein Menin, and the H3K79 methyltransferase DOT1L, which converges to inhibit myeloid differentiation by repressing a so-called "myeloid program" and maintaining the pro-tumorigenic leukemic identity of the KMT2A-r AML cells.
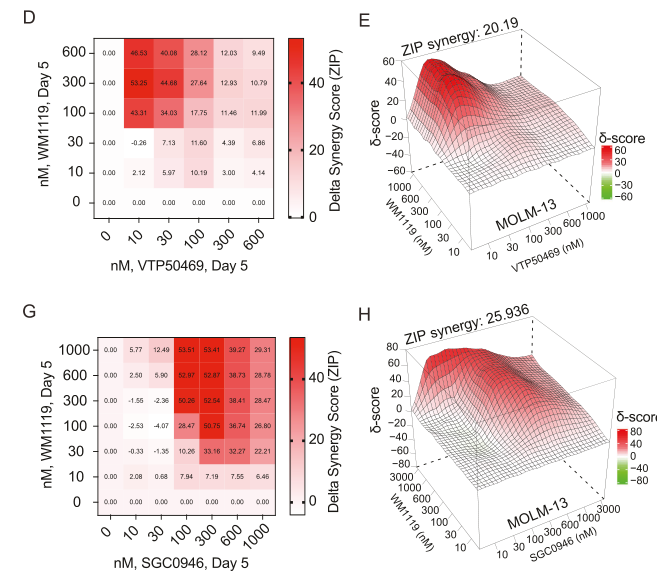
The perturbation of KAT6A, Menin, or DOT1L individually partially derepressed KMT2A-r AML cell differentiation; however, robust reactivation of differentiation and potent synergistic anti-leukemic activity required simultaneous pharmacological inhibition of these targets. This finding aligns with emerging models of "epigenetic cooperativity" in AML (Dafflon et al.), where multiple chromatin modifiers act in concert to enforce a stem-like cell fate. Furthermore, the therapeutic potential of this epigenetic co-dependency may also extend to reverse oncogenic gene expression programs in models of breast cancer (Olsen et al.).
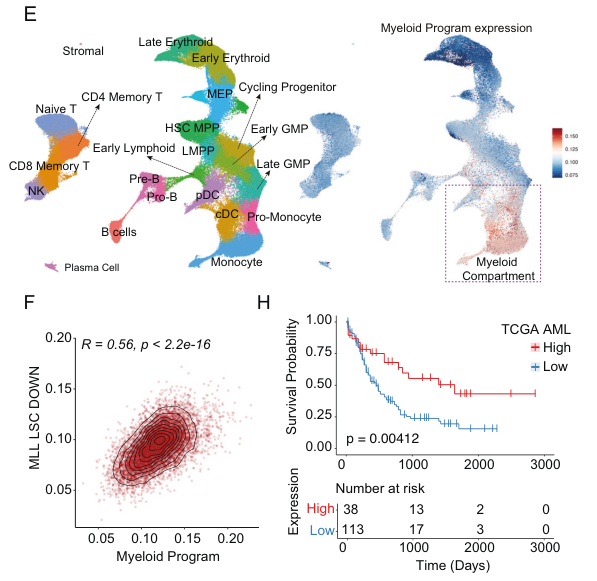
Of note, the authors also discovered an antagonistic relationship between DOT1L and the polycomb repressive complex 1.1 component PCGF1, with PCGF1 disruption conferring significant resistance to DOT1L inhibition. These findings provided a mechanistic explanation for therapeutic resistance, with previous studies reporting that PCGF1 loss confers resistance to Menin inhibitors (Zhou et al.) and highlight the need to disrupt multiple arms of a network to induce durable therapeutic responses.
Finally, the authors demonstrated the clinical relevance of the myeloid program, showing robust prognostic value in independent AML cohorts and predictive value for guiding therapeutic strategy.
Perturb-seq, Epigenetic Modifiers, and Acute Myeloid Leukemia: The Next Steps
Overall, this Perturb-seq-based study defined the epigenetic compensation mechanism that represses differentiation, sustains the leukemic state, and underlies therapeutic resistance, and, as such, provided a means to rationally design synergistic combination therapies for AML. Further studies reported as required by the authors include in vivo evaluations and primary patient studies, comprehensive efficacy and safety assessment in patient-derived xenograft models, mechanistic studies defining how Menin, KAT6A, and DOT1L assemble into a repressive module, and high-dimensional combinatorial perturbation strategies to explore the epigenetic landscape of this circuit more deeply.
The implementation of Paired-Tag technology from Epigenome Technologies, which generates joint epigenetic and transcriptomic profiles at single-cell resolution and detects histone modifications and RNA transcripts in individual nuclei with efficiency comparable to single-nucleus RNA-seq/ChIP-seq assays, has the potential to provide deeper insight into such research aims. Could the simultaneous single-cell analysis of histone modification and transcriptomic profiles help to provide more details regarding the epigenetic compensation mechanism underpinning therapeutic resistance in AML and other cancers?