Droplet Paired-Tag in Human Basal Ganglia
Can Droplet Paired-Tag Identify Cell-type Specific Cis-regulatory Elements (and More) in the Basal Ganglia? Droplet Paired-Tag supports the single-cell mapping of histone modification and transcriptome profiles across subregions of the human basal ganglia.
Droplet Paired-Tag: Single-Cell Mapping the Histone Modification and Transcriptome Profiles of the Basal Ganglia
Dysfunction of the basal ganglia, a group of interconnected subcortical nuclei in the brain that controls a variety of functions, contributes to the development of multiple neurological and psychiatric disorders. Single-cell transcriptomic studies have identified the neuronal and non-neuronal cell types present in rat and primate basal ganglia and their cell-type-specific gene expression profiles, which have provided deep insight into basal ganglia function and the mechanisms underlying related disease states. However, the gene regulatory programs that establish and maintain basal ganglia cell identity remain poorly defined.
The binding of transcription factors to cis-regulatory elements (CREs) - including promoters, enhancers, and silencers - constitutes the gene regulatory networks that control cell-type-specific gene expression profiles (Preissl et al.). Genetic variants in non-coding sequences associated with neurological and psychiatric disorders (Skene et al., Gaulton et al., and Frydas et al.) likely disrupt cell-specific CREs and alter target gene expression (Zhang et al.); however, the lack of data describing CREs and their activity in cells across brain regions represents a barrier to understanding regulatory mechanisms. As a solution, epigenomic profiling can help to annotate CREs, elucidate gene regulatory programs, and aid the interpretation of non-coding variants (Gaulton et al.). Previous studies that identified CREs in the human brain employed single-cell chromatin accessibility maps to catalog candidate CREs across brain cell types (Yi et al.); however, these approaches identified only active CREs and could not detect repressed or poised CREs. Swapping chromatin accessibility profiling for histone modification profiling may help to overcome this limitation; in addition, the dysregulation of histone modification profiles has been reported in cases of basal ganglia dysfunction (Borrelli et al.) and, as such, represents a basis for therapeutic interventions (Jones et al. and Nestler & Luscher).
Now, a new BioRxiv preprint from researchers led by Margarita Behrens and Bing Ren describes the application of the Droplet Paired-Tag technology from Epigenome Technologies (Xie et al.) to simultaneously profile the H3K27ac, H3K27me3, and H3K9me3 histone modifications and transcriptomes from the same single cells across multiple basal ganglia regions in neurotypical adult human donors to create the first single-cell multiome atlas of its kind. Paired-Tag technology from Epigenome Technologies generates joint epigenetic and transcriptomic profiles at single-cell resolution and detects histone modifications and RNA transcripts in nuclei with efficiency comparable to single-nucleus RNA-seq/ChIP-seq assays. Excitingly, the authors employ Droplet Paired-Tag to characterize active and repressive chromatin states at cell-type resolution, explore cell-type-specific gene regulatory programs, link non-coding neuropsychiatric risk variants to specific cell types, regulatory elements, and candidate target genes, and create a sequence-to-function deep-learning model that predicts gene regulation from DNA sequence and prioritizes functional disease-associated variants (Chang et al.). What can Droplet Paired-Tag technology and Epigenome Technologies do for your research? Let's find out.
Droplet Paired-Tag and the Basal Ganglia - All the Details!
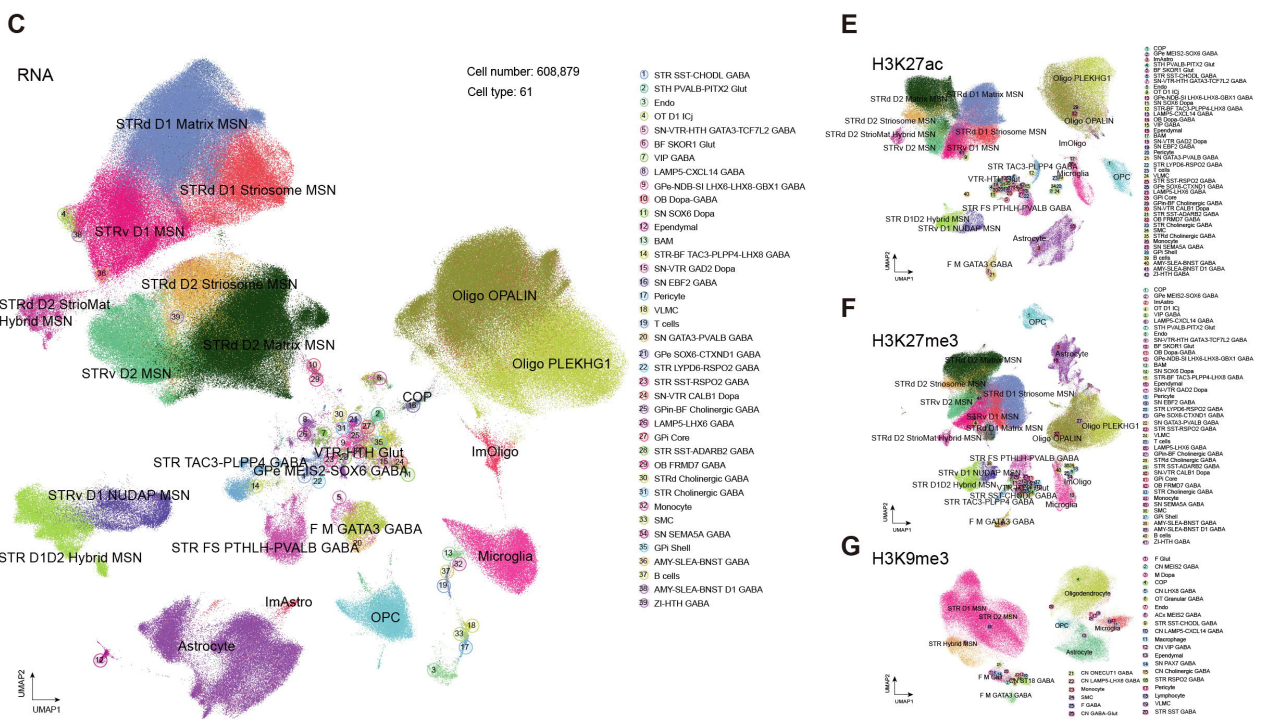
Expanding the Functional Epigenomic Landscape of the Basal Ganglia
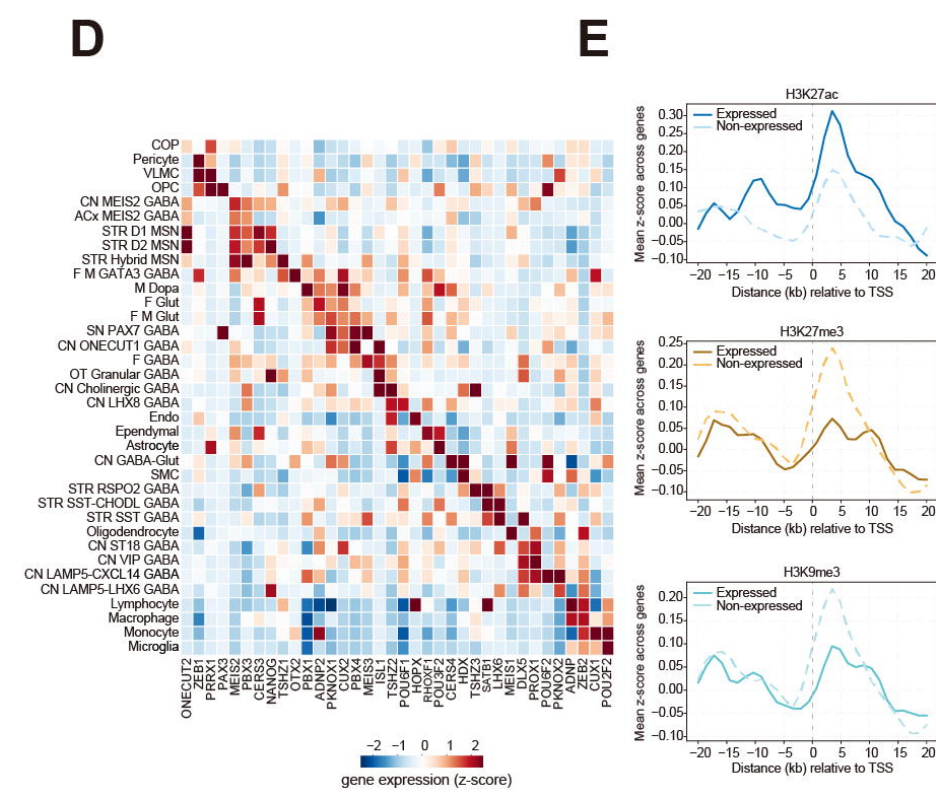
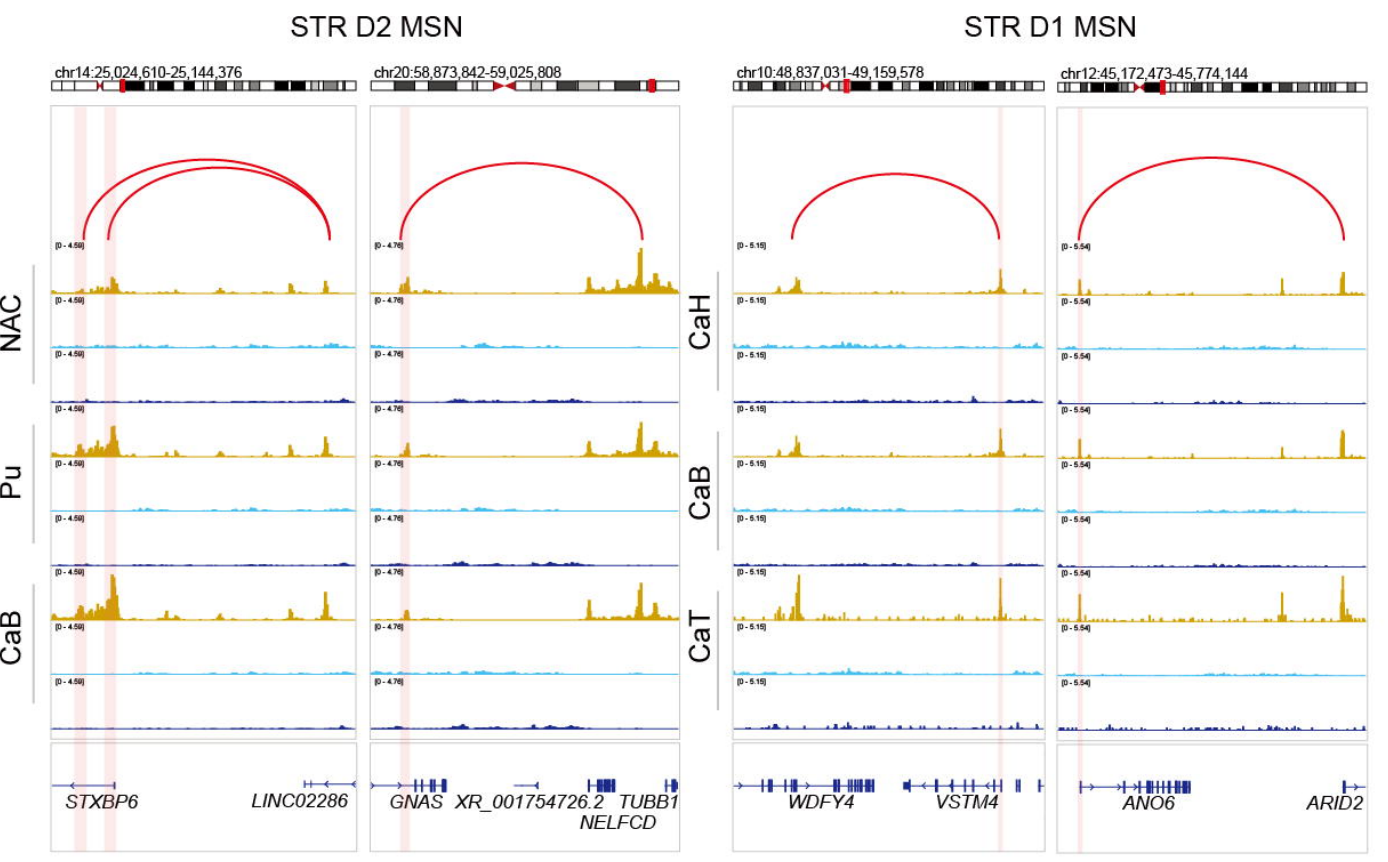
The analysis of human basal ganglia employed Droplet Paired-Tag to profile the transcriptome and histone modification landscapes (permissive modifications: H3K27ac; repressive modifications: H3K27me3 and H3K9me3) at single-nucleus resolution in samples dissected from eight anatomical subregions of seven neurotypical donor brains. The integration of histone modification and transcriptomic data from >600,000 single nuclei resolved 61 distinct basal ganglia cell types and annotated chromatin states covering approximately 50% of the genome; in comparison, previously published single-cell and bulk ATAC-seq profiles reached only 10%. Integrating and analyzing histone modification data with matching chromatin accessibility data revealed four active (displaying distinct chromatin accessibility and H3K27ac levels) and four repressive chromatin states (displaying distinct H3K9me3 and H3K27me3 levels). Subsequent in-depth analyses expanded the functional epigenomic landscape of human basal ganglia, identifying >530,000 CREs and potential target genes for >350,000; in addition, they identified ~180,000 repressive CREs that formed cell-type-specific loops with target genes and ~55,000 super-enhancers.
Integrating Spatial Data and Evaluating Evolutionary Conservation and Divergence
The authors also highlighted that integrating Droplet Paired-Tag data with additional datasets yielded interesting insights. The integration of a MERFISH-based spatial transcriptomic dataset (Chen et al.) helped to construct a high-resolution spatial epigenomic map that defined the organization of gene expression patterns and epigenomic regulation, uncovering regional heterogeneity in epigenomic landscapes in the basal ganglia. The authors discovered gradient changes in histone modifications at gene bodies and enhancers along anatomical basal ganglia axes, which paralleled transcriptomic gradients and, as such, provided the foundation for regional functional specialization. In addition, integrating human Droplet Paired-Tag data with a mouse brain Paired-Tag dataset enabled a systematic analysis of evolutionary conservation and divergence in the epigenome and gene regulation. The data indicated the broad conservation of medium spiny neurons group identities across species, particularly at promoter-proximal elements and core striatal identity modules that span active and repressive chromatin landscapes. Overall, the H3K27ac landscape displayed greater conservation than the H3K27me3 and H3K9me3 landscapes, due to the broader peak structure of repressive marks and their enrichment in transposable elements and related sequences.
Inferring Cell-Type-Resolved Regulatory Programs
What else could the authors understand from this Droplet Paired-Tag data? Excitingly, inferred cell-type-resolved regulatory programs revealed a combinatorial Homeobox transcription factor code that specified human neuronal cell-type identity and enabled the mechanistic interpretation of cell-type-specific gene expression signatures. The authors highlighted distinct regulatory programs for distinct types of medium spiny neuron subtypes in the basal ganglia, which exhibit highly similar transcriptomic and epigenomic states relative to other basal ganglia cell types. The distinctions between these two cell subtypes resided in the function of silencer elements identified from H3K27me3-based silencer-promoter loops (accompanied by subtype-specific transcription factor motif enrichment), which supported a model in which repressive chromatin stabilizes neuronal identity and constrains regulatory programs relevant to neuronal function. Furthermore, these data resolved epigenetic regulatory programs along the ventral-dorsal spatial axis and the developmental trajectories of oligodendrocytes in the human basal ganglia.
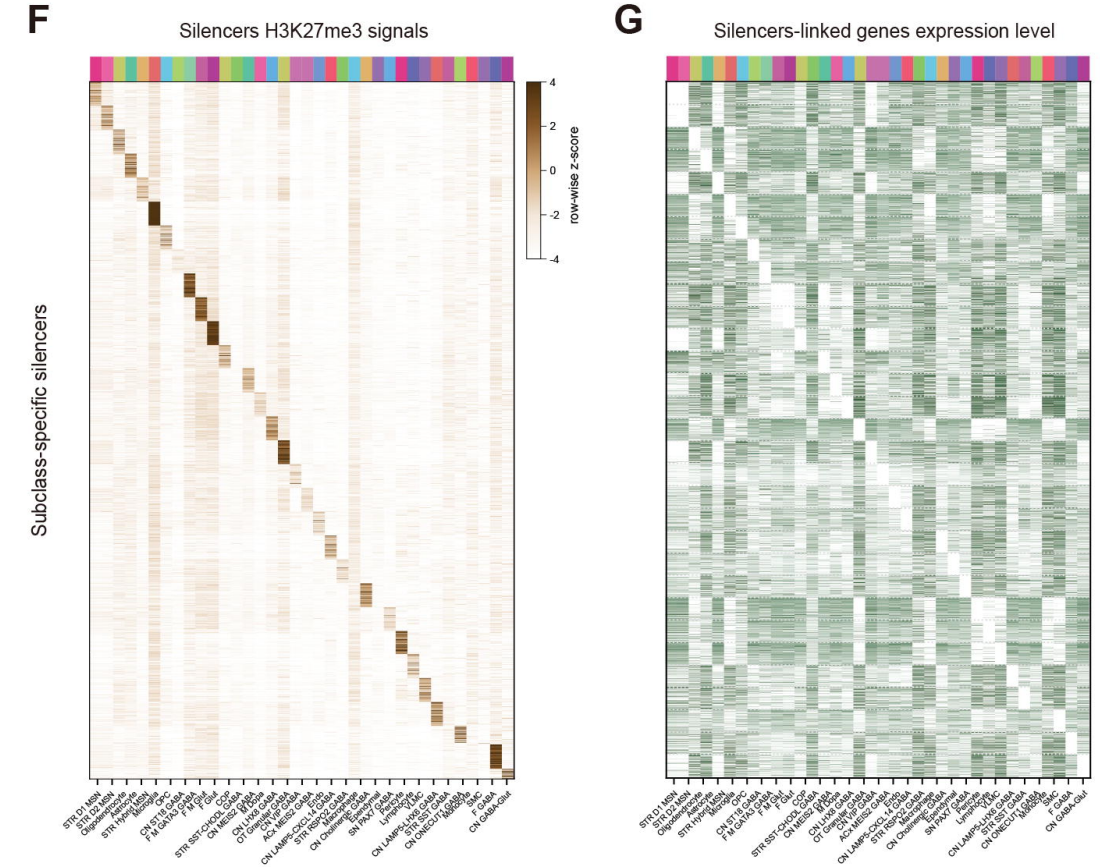
Exploring Non-coding Risk Variants in Basal Ganglia and Developing a Sequence-Based Deep-Learning Framework
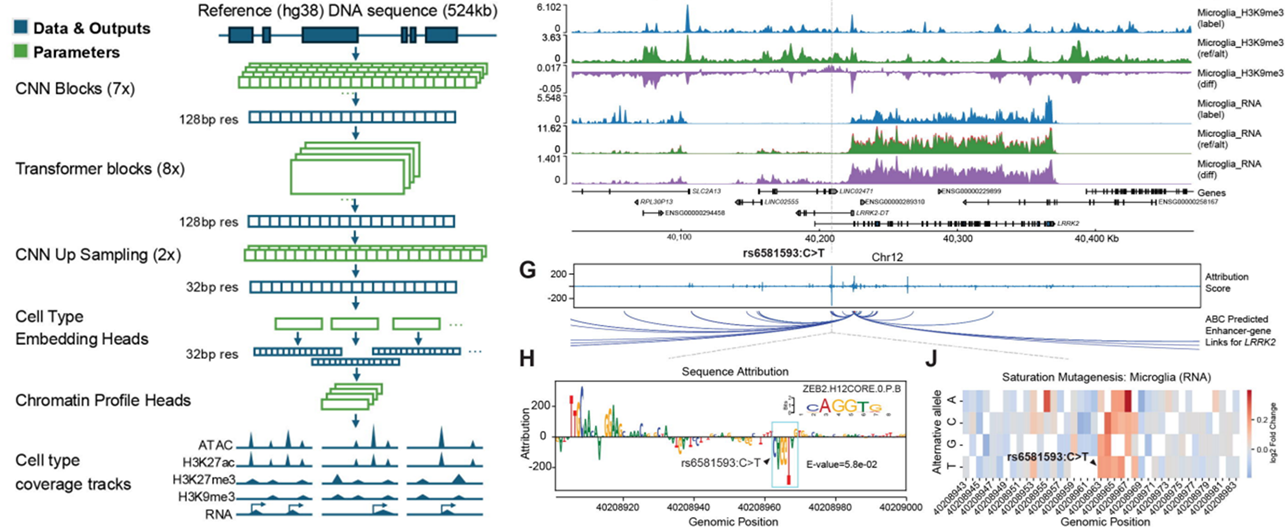
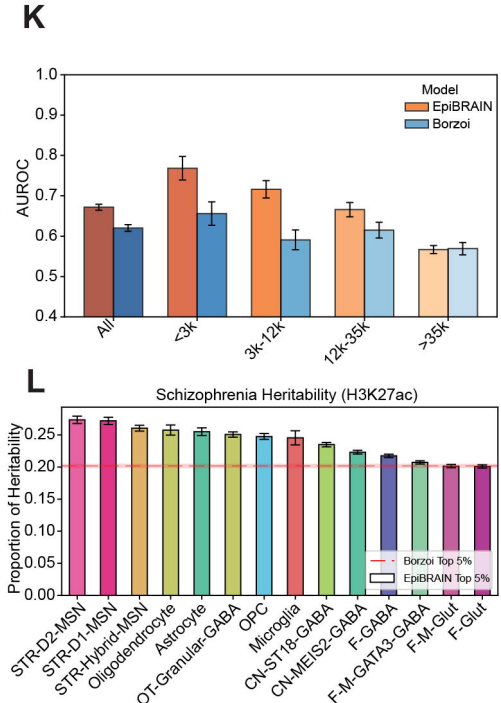
When seeking to interpret risk variants of complex traits and diseases, the study revealed how the cell-type-resolved histone modification landscapes and catalogs of CREs provided a framework for linking genome-wide association study variants to specific basal ganglia cell types, CREs, and putative target genes, thereby expanding mechanistic hypotheses and potentially identifying candidate therapeutic targets for neuropsychiatric disease. They discovered that H3K27ac peaks (but not H3K27me3 or H3K9me3 peaks) tended to display enrichment for Alzheimer’s disease risk variants in microglia and those for schizophrenia, intelligence, bipolar disorder, major depressive disorder, ADHD, neuroticism, insomnia, and tobacco in neurons.
Finally, the authors leveraged their Droplet Paired-Tag data to develop a sequence-based deep-learning framework called EpiBRAIN (Epigenomics-based Brain Regulation Attention Inference Network) (Johnson et al.), which predicted human brain cell-type-specific gene expression and multiple epigenomic profiles. Their results demonstrated how EpiBRAIN predicted epigenomic and transcriptional landscapes, identified sequence motifs that drove regulatory element function, and predicted the cell-type-specific effects of neuropsychiatric disease-associated non-coding variants. Encouragingly, EpiBRAIN also predicted the regulatory impact of a Parkinson’s disease risk variant and provided a novel mechanistic hypothesis. Overall, the results demonstrated that integrating histone modifications with sequence-based models enabled the in-silico interpretation of non-coding variants and provided a framework for prioritizing candidate regulatory mechanisms and therapeutic targets in neuropsychiatric disorders.
Droplet Paired-Tag and the Basal Ganglia - Concluding Comments and the Next Steps
This single-cell atlas of the human basal ganglia has expanded on chromatin accessibility-based CRE annotation and comprehensively mapped active and repressive epigenomic regulation by systematically annotating histone modification and transcriptomic profiles. These data provide a much deeper understanding of cell-type-specific gene regulatory programs and relevant non-coding disease risk variants, laying the groundwork for future investigations into the molecular underpinnings of neurological and psychiatric disorders in the basal ganglia. The authors do, however, note limitations to their study, which they aim to address through larger cohorts, improved protocols, and an expanded data scale.
For more information on epigenetic foundation models such as EpiBRAIN, see our previous post on Borzoi, AlphaGenome and other models; and for the fine details of how to fine-tune such models to establish a resource like EpiBRAIN, see our previous post on foundation model fine-tuning.
The implementation of Paired-Tag technology from Epigenome Technologies, which generates joint epigenetic and transcriptomic profiles at single-cell resolution and detects histone modifications and RNA transcripts in individual nuclei with efficiency comparable to single-nucleus RNA-seq/ChIP-seq assays, has the potential to provide deeper insight into such research aims. What more could the simultaneous single-cell analysis of histone modification and transcriptomic profiles tell us about other regions of the brain?